The simplest way to use Multi-Dendrix is to run the entire Multi-Dendrix pipeline. We have provided a program to run a customizable version of the Multi-Dendrix pipeline in the multi_dendrix_pipeline.py script. The analysis performed in the pipeline, the parameters for customizing the analysis, and documentation of the program itself are presented below.
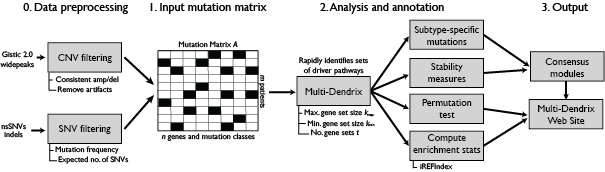
The Multi-Dendrix pipeline (implemented in multi_dendrix_pipeline.run()) consists of the following steps:
At this time, the pipeline does not include any functions for preprocessing mutation data.
The Multi-Dendrix pipeline accepts the following parameters. You may also be interested in the Input File Formats accepted by Multi-Dendrix.
| Name | Type | Description | Flag(s) | Required | Default | Example |
|---|---|---|---|---|---|---|
| Output directory | string | Directory name to store the text and HTML output. | -o --output_dir |
Yes. | N/A | "output" |
| Verbose | boolean | Flag whether to output progress or not. | -v --verbose |
No. | False | N/A |
| kmin | integer | Minimum gene set size. | -k_min --min_gene_set_size |
Yes. | N/A | 2 |
| kmax | integer | Maximum gene set size. | -k_max --max_gene_set_size |
Yes. | N/A | 3 |
| tmin | integer | Minimum number of gene sets. | -t_min --min_num_gene_sets |
Yes. | N/A | 2 |
| tmin | integer | Maximum number of gene sets. | -t_max --max_num_gene_sets |
Yes. | N/A | 3 |
| Data name | string | Name of data (for use in naming output files). | -n --db_name |
Yes. | N/A | "BRCA" |
| Mutation matrix file | string | Location of input mutation matrix. | -m --mutation_matrix |
Yes. | N/A | "BRCA.m2" |
| Mutation frequency cutoff | integer | Minimum number of patients a gene must be mutated in to be included in the mutation data. | -c --cutoff |
No. | 0 | 2 |
| Patient whitelist | string | See description of white- and blacklists. | -p --patient_whitelist |
No. | None | "BRCA.lst" |
| Patient blacklist | string | See description of white- and blacklists. | -bp --patient_blacklist |
No. | None | "BRCA.blst" | Gene whitelist | string | See description of white- and blacklists. | -g --gene_whitelist |
No. | None | "BRCA.glst" |
| Gene blacklist | string | See description of white- and blacklists. | -bg --gene_blacklist |
No. | None | "fishy_genes.glst" |
| α | float | Changes tradeoff between coverage and coverage overlap in the weight function used by Multi-Dendrix. | -a --alpha |
No. | 1.0 | 3.0 |
| Δ | integer | The number of overlaps allowed between gene sets in a single collection output by Multi-Dendrix. | -d --delta |
No. | 0 | 1 |
| λ | integer | The number of gene sets a gene is allowed to be a member in a single collection output by Multi-Dendrix. | -l --lmbda |
No. | 1 | 2 |
| Stability threshold for core modules | integer | The number of gene sets a pair of genes must be a member of together to be grouped into the same core module. | --stability_threshold | No. | 2 | 1 |
| Perform subtype analysis | boolean | Flag whether the pipeline should include subtype-specific mutation analysis. | --subtypes | No. | False | N/A |
| Significance level for subtype-specific mutations | float | The maximum Bonferonni-corrected p-value for reported subtype-specific mutations. | --subtype_sig_threshold | No. | 0.05 | 0.01 |
| Perform network test | boolean | Flag whether to perform the network permutation test. | --network_test | No. | False | N/A |
| Input PPI network | string | File location of an input PPI network (formatted as a NetworkX edge list). | -ppi --network_edgelist |
No. | None | "iref_edgelist" |
| Number of permuted networks | integer | Number of permuted PPI networks to create when performing the direct interactions test. Only used when a directory location of premuted networks is not provided. | --num_permuted_networks | No. | 5 | 1000 |
| Permuted networks directory | string | Directory location of permuted network edge lists. | --permuted_networks_dir | No. | None | "permuted_iref/" |
| Average pairwise distance flag | boolean | Flag whether to perform the average pairwise distance test instead of the direct interactions test. | --distance | No. | False | N/A |
| Q | integer | Parameter to control the number of edge swaps when permuting PPI networks. For graph G=(V, E), Q * |E| edge swaps are performed. | --Q | No. | 100 | 5 |
| Perform statistical significance test | boolean | Flag whether to perform the statistical significance test. | --weight_test | No. | False | N/A |
| Number of permuted matrices | integer | Number of permuted matrices to create when calculating statistical significance. Only used when a directory location of premuted matrices is not provided. | --num_permuted_matrices | No. | 5 | 100 |
| Permuted matrices directory | string | Directory location of permuted matrices. | --permuted_matrices_dir | No. | None | "permuted_BRCA/" |
| run(args) | Runs the whole Multi-Dendrix Pipeline for the given command-line arguments. |
| batch_multi_dendrix(args) | Runs Multi-Dendrix for each parameter setting on the input |
| run_network_permutation_test(args, ...) | Runs the direct interactions or average pairwise distance test on each of the collections and the core_modules. |
| run_matrix_permutation_test(args, ...) | Runs the direct interactions or average pairwise distance test on each of the collections and the core_modules. |