![]()
![]()
HATCHet¶
HATCHet is an algorithm to infer allele and clone-specific copy-number aberrations (CNAs), clone proportions, and whole-genome duplications (WGD) for several tumor clones jointly from multiple bulk-tumor samples of the same patient or from a single bulk-tumor sample. HATCHet has been designed and developped by Simone Zaccaria in the group of prof. Ben Raphael at Princeton University. The full description of the algorithm, the comparison with previous state-of-the-art methods, and its application on published cancer datasets are described in
Simone Zaccaria and Ben Raphael, 2018
The novel simulationg framework, MASCoTE, is available at
The simulated data, the results of all the methods considered in the comparison, the results of HATCHet on the published whole-genome multi-sample tumor sequencing datasets ((Gundem et al., Nature, 2015) and (Makohon-Moore et al., Nature genetics, 2017)) and all the related analyses are available at
This repository includes a quick overview of the HATCHet’s algorithm and software, detailed instructions for installation and requirements, a script to execute the full pipeline of HATCHet, demos to illustrate how to apply HATCHet on different datasets with different features, a list of current issues, and contacts.
Contents¶
Overview¶
Algorithm¶
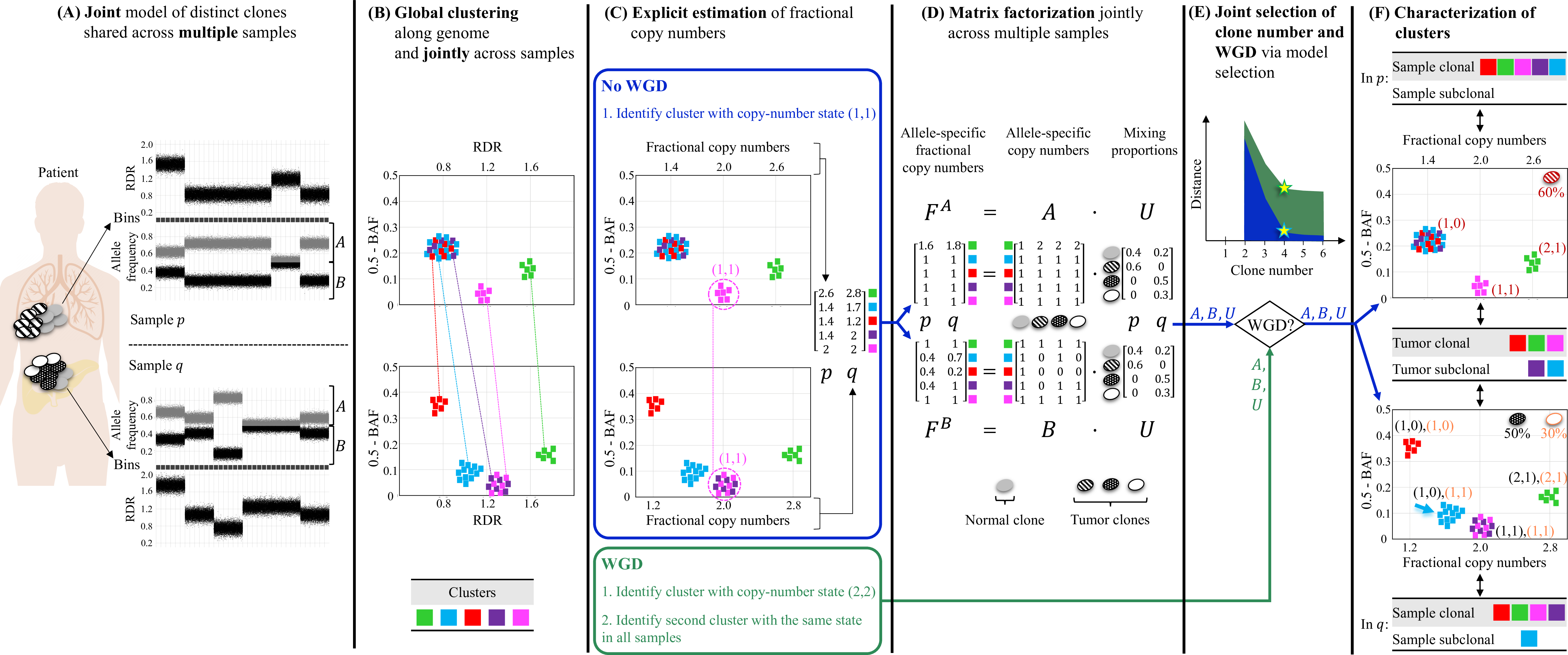
Overview of HATCHet algorithm.
HATCHet analyzes the read-depth ratio (RDR) and the B-allele frequency (BAF) in bins of the reference genome (black squares) jointly from multiple tumor samples. Here, we show two tumor samples p and q.
HATCHet globally clusters the bins based on RDR and BAF along the entire genome and jointly across samples p and q. Each cluster (color) includes bins with the same copy-number state within each clone present in p or q.
HATCHet estimates the fractional copy number of each cluster. If there is no WGD, the identification of the cluster (magenta) with copy-number state (1, 1) is sufficient and RDRs are scaled correspondingly. If a WGD occurs, HATCHet finds the cluster with copy-number state (2, 2) (same magenta cluster) and a second cluster having an identical copy-number state in all tumor clones.
HATCHet factorizes the allele-specific fractional copy numbers F^A, F^B into the allele-specific copy numbers A, B, respectively, and the clone proportions U. Here there is a normal clone and 3 tumor clones.
HATCHet’s model selection criterion identifies the matrices A, B and U in the factorization while evaluating the fit according to both the inferred number of clones and presence/absence of a WGD.
Clusters are classified by their inferred copy-number states in each sample. Sample-clonal clusters have a unique copy-number state in the sample and correspond to evenly-spaced positions in the scaled RDR-BAF plot (vertical grid lines in each plot). Sample-subclonal clusters (e.g. cyan in p) have different copy-number states in a sample and thus correspond to intermediate positions in the scaled RDR-BAF plot. Tumor-clonal clusters have identical copy-number states in all tumor clones – thus they are sample-clonal clusters in every sample and preserve their relative positions in scaled-RDR-BAF plots. In contrast, tumor-subclonal clusters have different copy-number states in different tumor clones and their relative positions in the scaled RDR-BAF plot varies across samples (e.g. purple cluster).
Note that this overview and figure do not include recently added features such as variable-width binning and locality-aware clustering that are currently default in HATCHet. These features will be described in a future publication.
Software¶
The current implementation of HATCHet is composed of two sets of modules:
(1) The core modules of HATCHet are designed to efficiently solve a challenging constrained and distance-based simultaneous matrix factorization which aim to infer allele and clone-specific copy numbers and clone proportins from fractional copy numbers. The module is implemented in C++11 and are included in src folder.
(2) The utility modules of HATCHet perform several different tasks that are needed to process the raw data, perform steps of the HATCHet’s algorithm needed for the factorization, and process the results. These task include reading/calling germinal single-point mutations, counting reads from a BAM file, combining the read counts and other information, segmenting through HATCHet’s global approach, plotting very useful information, etc. These modules are implemented in python 3 and are available as the util and bin submodules.
Setup¶
The setup process is composed of 3 steps:
Installation: the compilation and installation of Hatchet and its dependencies; this is a one-time process.
Using a Solver: the requirements to run a Solver; these need to be satisfied for computing allele-specific fractional copy numbers.
Required data: the requirements for considered data; these need to be satisfied whenever using new data.
Installation¶
Standard Installation¶
HATCHet can be installed using an existing installation of conda (e.g. either the compact
Miniconda or the complete Anaconda).
We recommend creating a new environment in which to install hatchet:
conda create -n hatchet hatchet
In this case, hatchet must be activated before every session with the following command
source activate hatchet
For installation, bioconda requires to set the following channels in this exact order:
conda config --add channels defaults
conda config --add channels bioconda
conda config --add channels conda-forge
Then, hatchet can be installed with the following one-time command:
conda install hatchet
If you would like to run the reference-based phasing modules of HATCHet, please install the additional dependency shapeit from the channel dranew:
conda install -c dranew shapeit
Manual Installation¶
If you wish to install HATCHet directly from this repository, the steps are a bit more involved. Please refer to the
Manual Installation document for more details.
Using a Solver¶
Every run of HATCHet (specifically, the compute-cn step) needs to use a Pyomo supported solver. By default, the HATCHet is compiled against Gurobi, so the easiest (and fastest) option is to use a valid Gurobi license.
Using Gurobi¶
We recommend using the latest version of Gurobi. For a full list of compatible Python and Gurobi versions, see the Python/Gurobi compatibility table.
If using Gurobi (the default option), make sure that the environmental variable GRB_LICENSE_FILE points to a valid license file (typically with a .lic extension). This can be easily obtained depending on the type of free academic license available:
Individual license. This license can be obtained easily by any academic user with an institutional email. This license is user and machine-specific, meaning that the user needs to require a different license for every used machine. Assuming the license is stored at
/path/to/gurobi.lic, set the environment variableGRB_LICENSE_FILEto point to it:export GRB_LICENSE_FILE="/path/to/gurobi.lic"
Multi-use license. This license can be used by multiple users on any machine in a cluster. This license can be obtained but needs to be requested by the IT staff of the user’s institution. This license is typically used in a machine cluster and only requires the following command (the exact form of this command may vary):
module load gurobi
Using a different Pyomo-supported solver¶
If you do not have or wish to use Gurobi (though using Gurobi will be at least twice as fast as any other solver),
you can use any Pyomo-supported solver by setting the environment variable HATCHET_COMPUTE_CN_SOLVER to cbc,
glpk, or any other Pyomo-supported solver. For example, export HATCHET_COMPUTE_CN_SOLVER=cbc.
Alternatively, you can set the key solver key in the compute_cn
section of your hatchet.ini (if using the hatchet run command) to a specific Pyomo-supported
solver. Make sure the relevant solver binaries are in your $PATH, otherwise Pyomo will not be able to find them
correctly.
One HATCHet command that is very useful to sanity-check your solver is hatchet check. This command runs the HATCHet compute_cn step on a small set of pre-packaged data files and completes fairly quickly (a few seconds for the Gurobi optimizer, but up to a few minutes for glpk). Running this command will ensure that you have your solver settings (including licenses) set up correctly, so you should always run this command first before trying out the compute_cn step on your large data files.
Required data¶
HATCHet requires 3 input data files:
One or more BAM files containing DNA sequencing reads obtained from tumor samples of a single patient. Every BAM file contains the sequencing reads from a sample ad needs to be indexed and sorted. For example, each BAM file can be easily indexed and sorted using SAMtools. In addition, one can improve the quality of the data by processing the BAM files according to the GATK Best Practices.
A BAM file containg DNA sequencing reads obtained from a matched-normal sample of the same patient of the considered tumor samples. The BAM file needs to be indexed and sorted as the tumor BAM files. Also, the BAM files can be processed as the tumor BAM files.
A human reference genome. Ideally, one should consider the same human reference genome used to align the sequencing reads in the given BAM files. The most-used human reference genome are available at GRC or UCSC. Observe that human reference genomes use two different notations for chromosomes: either
1, 2, 3, 4, 5 ...orchr1, chr2, chr3, chr4, chr5 .... One needs to make sure all BAM files and reference genome share that same chromosome notation. When this is not the case, one needs to change the reference to guarantee consistency and needs to re-index the new reference (e.g. using SAMtools). Also, HATCHet requires that the name of each chromosome is the first word in each ID such that>1 [ANYTHING] ... \n>2 [ANYTHING] ... \n>3 [ANYTHING] ...or>chr1 [ANYTHING] ... \n>chr2 [ANYTHING] ... \n>chr3 [ANYTHING].For the reference genome, HATCHet requires the existence of a a sequence dictionary (
.dict), which is part of all standard pipelines for sequencing data, see for example GATK or Galaxy. Please note that the sequence dictionary is NOT the reference index.fai, which is a different structure, has a different function, and it is also recommended.The dictionary of a reference genome is often included in the available bundles for the reference genomes, see the example for hg19 from Broad Institute. However, the dictionary can also be generated in seconds using either SAMtools or Picard tools.
In the folder where you want to download and index the human genome, the steps would typically be:
curl -L https://hgdownload.soe.ucsc.edu/goldenPath/hg19/bigZips/hg19.fa.gz | gzip -d > hg19.fa samtools faidx hg19.fa samtools dict hg19.fa > hg19.dict
Usage¶
The repository includes all the components that are required to cover every step of the entire HATCHet’s pipeline, starting from the processing of raw data reported in a BAM file through the analysis of the final results. We provide:
A script representing the full pipeline of HATCHet, and we describe in details the whole script through a tutorial with instructions for usage.
Demos that correspond to guided executions of HATCHet on some examples, and explain in detail the usage of HATCHet when considering standard datasets, real datasets with high noise, and different kind of data.
Custom pipelines which adapt the full HATCHet’s pipeline to special conditions or integrates pre-processed data belonging to different pipelines.
The implementation of HATCHet is highly modular and one can replace any HATCHet’s module with any other method to obtain the required results (especially for the pre-processing modules). As such, we also provide here an overview of the entire pipeline and we describe the details of each step in a dedicated section of the manual.
Recommendations, especially for noisy datasets or with different features, to guide the user in the interpretation of HATCHet’s inference. We explain how to perform quality control to guarantee the best-quality results, and describe how the user can control and tune some of the parameters to obtain the best-fitting results.
Current issues¶
HATCHet is in active development, please report any issue or question as this could help the devolment and imporvement of HATCHet. Current known issues with current version are reported here below:
The allele-swapping feature of combine-counts has been temporarily disabled due to conflicts with recent SAMtools versions.
HATCHet has not been tested on Windows yet. For Windows users, we recommend Windows Subsystems for Linux.
A list of the major recent updates:
Variable-width binning which adapts to the sequencing coverage and observed heterzygous germline SNP positions
Modules for reference-based phasing and associated pre- and post-processing
Locality-aware clustering which combines global clustering with genomic location
Added plotting modules which show a) bins with consistent coloring between 2D and genome-wide views and b) expected positions of assigned copy-number states
Contacts¶
HATCHet is maintained by the research groups of Prof. Ben Raphael (Princeton) [email] and Prof. Simone Zaccaria (UCL) [email]. Major contributors include Matt Myers, Vineet Bansal, and Brian Arnold.