Demo for standard WGS data¶
: ex: set ft=markdown ;:<<’```shell’ #
NOTE: this demo has not yet been updated for version 1.0 of HATCHet which includes variable-width binning, phasing, and locality-aware clustering.
The following HATCHet’s demo represents a guided example starting from WGS (whole-genome sequencing) data from 3 simulated samples obtained from the same tumor. This represents an exemplary case of executing HATCHet on a typical dataset with standard noise. For simplicity, the demo starts from a BB file demo-WGS-sim.bb (included in this demo at examples/demo-WGS-sim/) which contains the RDR and BAF of every genomic bin and, therefore, we assume that the preliminary steps (i.e. count-reads, count-alleles, and combine-counts) have already been executed by running standard configuration for WGS data (bin size of 50kb through -b 50kb of count-reads, and the allele counts for germline heterozygous SNPs have been selected between 3 and 200 through -c 3 -C 200).
Specifically, the tumor is composed of 2 tumor clones (clone0 and clone1) and a normal diploid clone (normal). The clonal composition of every sample is described in the following table:
| Name | Proportion of normal clone | Proporion of clone0 | Proportion of clone1 |
|---|---|---|---|
bulk_035clone1_06clone0_005normal |
5% |
60% |
35% |
bulk_055clone1_04clone0_005normal |
5% |
40% |
55% |
bulk_08clone1_Noneclone0_02normal |
20% |
Not present | 80% |
Requirements and set up¶
The demo requires that HATCHet has been successfully compiled and all the dependencies are available and functional. As such, the demo requires the user to properly set up the following paths:
PY="python3" # This id the full path to the version of PYTHON3 which contains the required `hatchet` module. When this corresponds to the standard version, the user can keep the given value of `python3`
:<<'```shell' # Ignore this line
The following paths are consequently obtained to point to the required components of HATCHet
CLUSTERBINS="${PY} -m hatchet cluster-bins"
PLOTBINS="${PY} -m hatchet plot-bins"
INFER="${PY} -m hatchet compute-cn"
PLOTCN="${PY} -m hatchet plot-cn"
:<<'```shell' # Ignore this line
We also ask the demo to terminate in case of errors and to print a trace of the execution by the following commands
set -e
set -o xtrace
PS4='[\t]'
:<<'```shell' # Ignore this line
Global clustering¶
The first main step of the demo performs the global clustering of HATCHet where genomic bins which have the same copy-number state in every tumor clone are clustered correspondingly. To do this, we use cluster-bins, i.e. the HATCHet’s component designed for this purpose. At first, we attempt to run the clustering using the default values of the parameters as follows:
${CLUSTERBINS} demo-wgs-sim.bb -o demo-wgs-sim.seg -O demo-wgs-sim.bbc -e 12 -tB 0.03 -tR 0.15 -d 0.08
:<<'```shell' # Ignore this line
To assess the quality of the clustering we generate the cluster plot using the CBB command of plot-bins, i.e. the HATCHet’s component designed for the analysis of the data. For simplicity, we also use the following options
--colwrap 3allows to have the 3 plots of the 3 samples on the same figure row-tS 0.01asks to plot only the clusters which cover at least the1%of the genome. This is useful to clean the figure and focus on the main components.
${PLOTBINS} -c CBB demo-wgs-sim.bbc --colwrap 3 -tS 0.01
:<<'```shell' # Ignore this line
We thus obtain the following clustering:
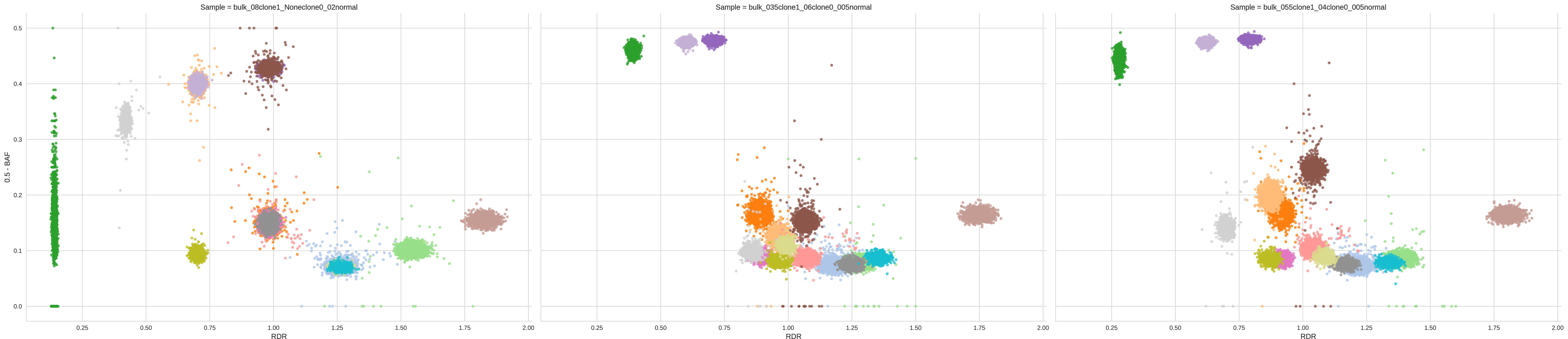
We can easily notice that the clustering is good and not tuning is needed as every pair of clusters is clearly distinct in one of the two dimensions (RDR and BAF) in at least one sample.
hatchet’s step¶
Next we apply hatchet, i.e. the component of HATCHet which estimates fractional copy numbers, infers allele-and-clone specific copy numbers, and jointly predicts the number of clones (including the normal clone) and the presence of a WGD.
We apply the last step with default parameters and, for simplicity of this demo, we consider 8 clones, which can be easily considered by HATCHet in this case, and we only consider 100 restarts for the coordinate-descent method; these are the number of attempts to find the best solution. This number is sufficient in this small example but we reccommend to use at least 400 restarts in standard runs.
${INFER} -i demo-wgs-sim -n2,8 -p 100 -v 2 -u 0.03 -r 12 -eD 6 -eT 12 -l 0.5 |& tee hatchet.log
:<<'```shell' # Ignore this line
We obtain the following summary of results:
## Scores approximating second derivative for diploid results
## Diploid with 2 clones - OBJ: 66.884758 - score: -0.17739731674
## Diploid with 3 clones - OBJ: 34.954154 - score: -0.0216588755513
## Diploid with 4 clones - OBJ: 17.510067 - score: 0.0555719383474
## Diploid with 5 clones - OBJ: 9.744628 - score: 0.186776250313
## Diploid with 6 clones - OBJ: 7.243104 - score: 0.0839737725603
## Diploid with 7 clones - OBJ: 5.991972 - score: 0.0538665194051
## Diploid with 8 clones - OBJ: 5.27972 - score: -0.181132288335
## Scores approximating second derivative for tetraploid results
## Tetraploid with 2 clones - OBJ: 81.216417 - score: -0.415141225696
## Tetraploid with 3 clones - OBJ: 23.135209 - score: 0.169099207187
## Tetraploid with 4 clones - OBJ: 15.47975 - score: 0.0653131702741
## Tetraploid with 5 clones - OBJ: 11.36852 - score: 0.0682096876804
## Tetraploid with 6 clones - OBJ: 9.124625 - score: 0.0411292123085
## Tetraploid with 7 clones - OBJ: 7.698914 - score: 0.0753487410723
## Tetraploid with 8 clones - OBJ: 7.076072 - score: -0.219100018522
# The chosen diploid solution has 5 clones with OBJ: 9.744628 and score: 0.186776250313
## The related-diploid resulting files are copied to ./chosen.diploid.bbc.ucn and ./chosen.diploid.seg.ucn
# The chosen tetraploid solution has 3 clones with OBJ: 23.135209 and score: 0.169099207187
## The related-tetraploid resulting files are copied to ./chosen.tetraploid.bbc.ucn and ./chosen.tetraploid.seg.ucn
# The chosen solution is tetraploid with 3 clones and is written in ./best.bbc.ucn and ./best.seg.ucn
HATCHet predicts the presence of 3 clones in the 3 tumor samples and, especially, predicts that 2 samples contains two distinct tumor clones, according to the true clonal composition.
Analyzing inferred results¶
Finally, we obtain useful plots to summarize and analyze the inferred results by using plot-cn, which is the last component of HATCHet. We run plot-cn as follows
${PLOTCN} best.bbc.ucn
exit $?
First, plot-cn summarizes the values of tumor purity and tumor ploidy for every sample of the patient as follows:
### SAMPLE: bulk_08clone1_Noneclone0_02normal -- PURITY: 0.799477 -- PLOIDY: 3.07107541299 -- CLASSIFICATION: TETRAPLOID
### SAMPLE: bulk_035clone1_06clone0_005normal -- PURITY: 0.95065 -- PLOIDY: 3.37447061915 -- CLASSIFICATION: TETRAPLOID
### SAMPLE: bulk_055clone1_04clone0_005normal -- PURITY: 0.95048 -- PLOIDY: 3.27575289096 -- CLASSIFICATION: TETRAPLOID
Next, plot-cn produces some informative plots to evaluate the inferred results. Among all the plots, 3 of those are particularly interesting.
The first intratumor-clones-totalcn.pdf represents the total-copy numbers for all tumor clones in fixed-size regions (obtained by merging neighboring genomic bins).
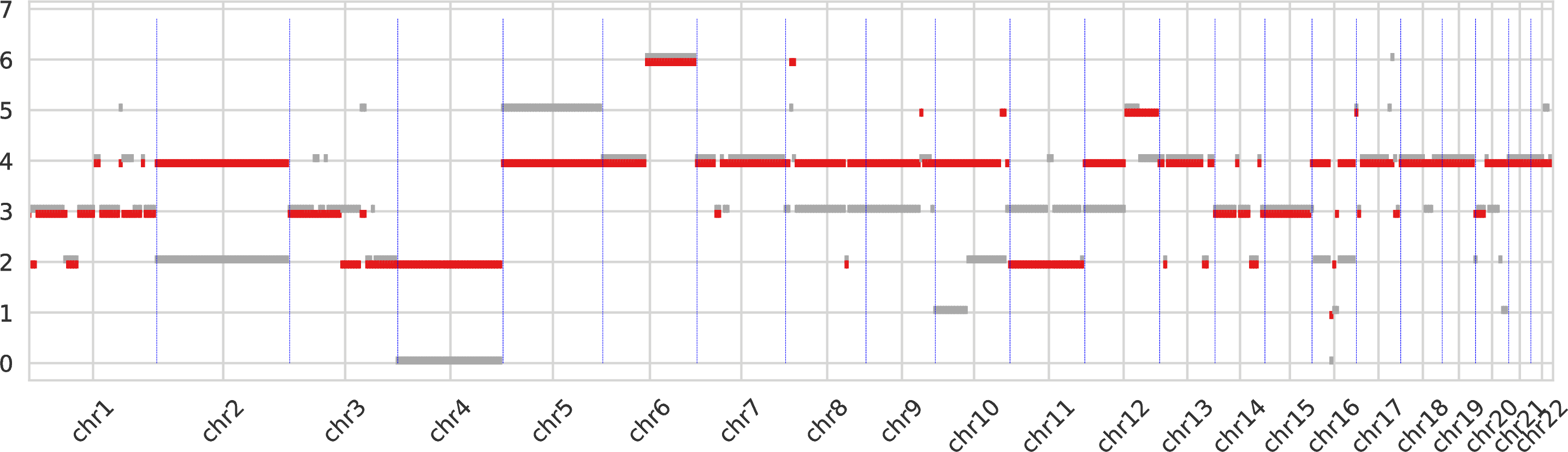 Every tumor clone is identified by a color and a dot is drawn for every genomic region (which are partitioned) for the corresponding total copy number.
Every tumor clone is identified by a color and a dot is drawn for every genomic region (which are partitioned) for the corresponding total copy number.
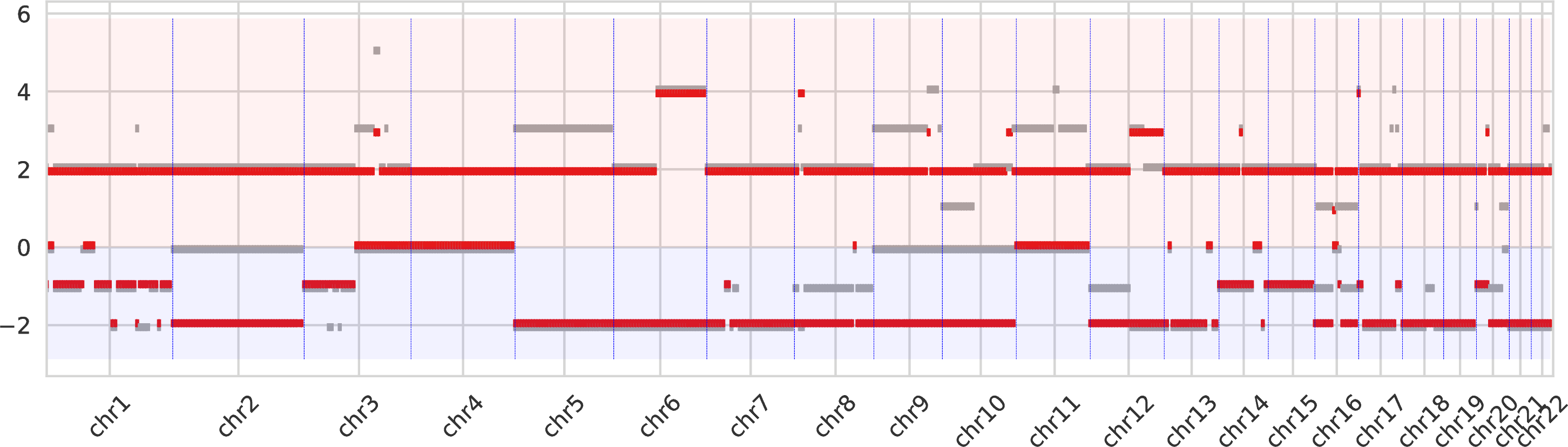
The second intratumor-clones-allelecn.pdf similarly represents the allele-specific copy numbers (split between the bottom and top regions of the figure) as the plot above.
The third intratumor-profiles.pdf represents both the clone proportions and the total copy numbers of every clone in genomic regions.
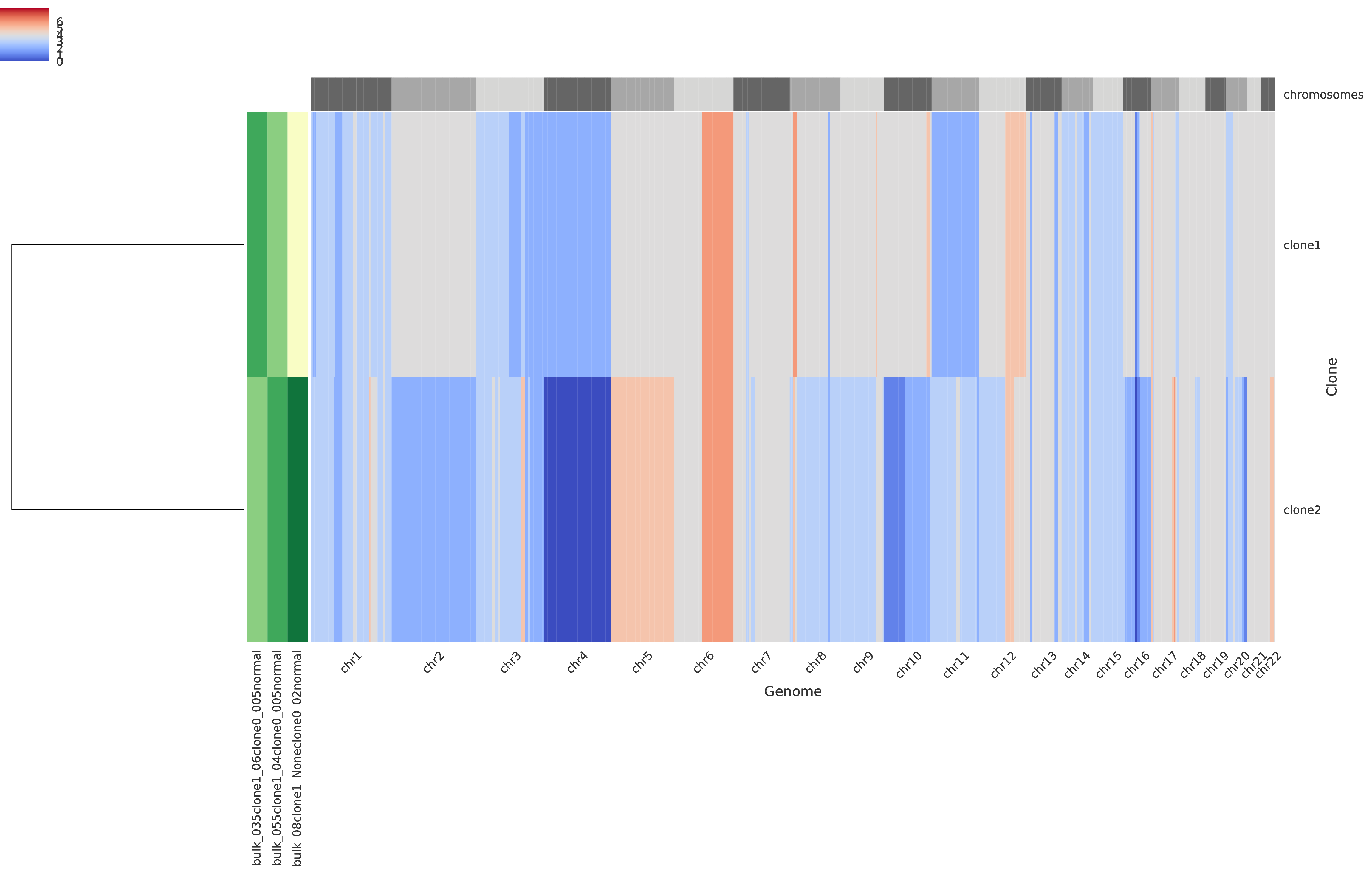 The main heatmap in the right side of the figure represent the total copy number of every clone (i.e. a row) for every genomic region (which are partition into chromosomes as described at the top of the heatmap) such that grey color indicate regions with base copy number not affected by CNAs (but they can be affected by a WGD when this occurrs), blue colors indicates deletions, and red colors indicate amplifications; in general, the stronger the color the smaller/higher the corresponding aberration. The smaller heatmap in the left side describes the clone proportion of each clone in each sample such that the lightest color correspond to absence while darker colors indicate higher clone proportions. Last, in the left-most part there is a dendogram which hierarchically clusters the tumor clones based on similiraity.
The main heatmap in the right side of the figure represent the total copy number of every clone (i.e. a row) for every genomic region (which are partition into chromosomes as described at the top of the heatmap) such that grey color indicate regions with base copy number not affected by CNAs (but they can be affected by a WGD when this occurrs), blue colors indicates deletions, and red colors indicate amplifications; in general, the stronger the color the smaller/higher the corresponding aberration. The smaller heatmap in the left side describes the clone proportion of each clone in each sample such that the lightest color correspond to absence while darker colors indicate higher clone proportions. Last, in the left-most part there is a dendogram which hierarchically clusters the tumor clones based on similiraity.